对25,000多个原子进行纳秒级MD模拟,DeepMind开发基于ML的大规模分子模拟通用方法
 发布于2024-12-26 阅读(0)
发布于2024-12-26 阅读(0)
扫一扫,手机访问

编辑 | 萝卜皮
深入了解复杂的过程是分子动力学(MD)模拟可以做到的,但准确的MD模拟需要昂贵的量子力学计算。对于较大的系统,使用高效但不太可靠的经验力场。机器学习力场(MLFF)提供与从头计算方法相当的精度,速度更快更高效,但难以模拟大分子中的长程相互作用。
Google DeepMind、柏林工业大学(Technische Universität Berlin)和卢森堡大学(University of Luxembourg)的研究人员提出了一种通用方法 GEMS,通过对「自下而上」和「自上而下」分子片段进行训练,来构建用于大规模分子模拟的准确 MLFF。
GEMS+允许以从头计算级别的质量,对超过25,000个原子进行纳米级MD模拟,正确预测聚丙烯酸丙酯中不同螺旋基序之间的动态振动,并与溶剂化Crambin 中大规模蛋白质-水波动的太赫兹光谱产生良好的一致性。该团队分析表明,从头开始精度的模拟对于理解动态生物分子过程是必要的。
研究以「Biomolecular dynamics with machine-learned quantum-mechanical force fields trained on diverse chemical fragments」为题,于 2024 年 4 月 5 日发布在《Science Advances》。

分子动力学(MD)模拟通过计算化学和生物过程中单个原子的运动,为理解分子属相和功能提供了解释。
然而,由于精确求解多体薛定谔方程的难度,目前仅适用于短期内少量原子的模拟,而近似的经验力场(FF)则因其计算效率而广泛应用于较大系统的模拟。
在需要额外准确性和灵活性的情况下,量子力学/分子力学(QM/MM)模拟提供了一种替代方案,即将系统分为用量子力学方法建模的小QM区域和用经典力学描述的大MM区域。
近年来,机器学习力场(MLFF)作为一种新的 MD 模拟手段出现,它结合了传统 FF 的计算效率和量子化学方法的高精度,通过在从头算参考数据上训练机器学习模型来预测能量和力,无需显式求解薛定谔方程。
虽然在小到中等规模系统的模拟中,MLFF 取得了成功,但构建适用于大型异质系统(如蛋白质或其他生物相关系统)的 MLFF 仍面临挑战。
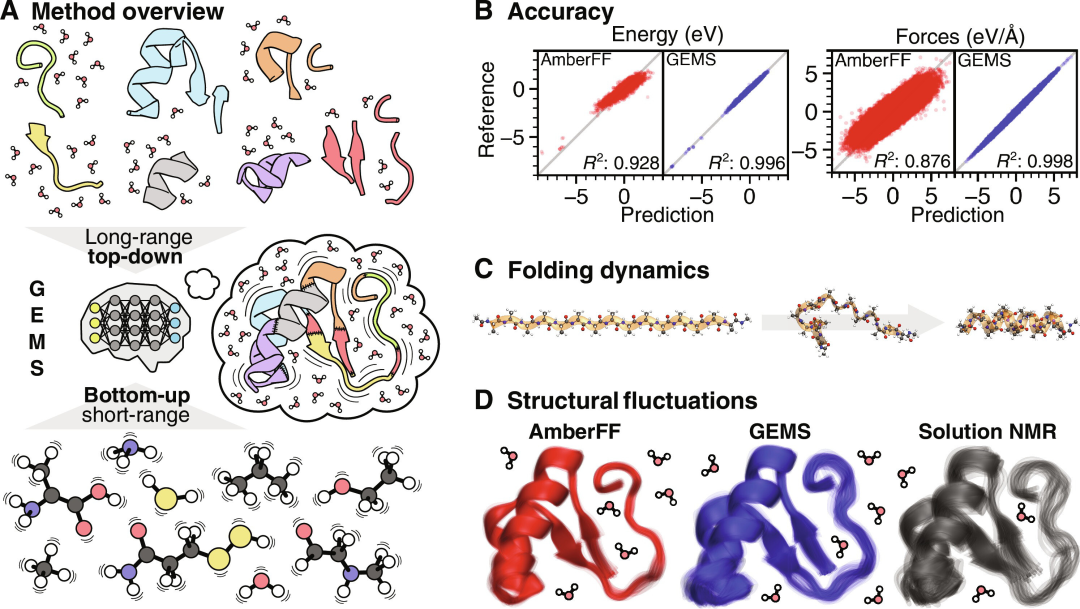
在最新的工作中,Google DeepMind 和卢森堡大学的研究人员提出了一种为大规模分子模拟构建精确 MLFF 的通用方法,称为 GEMS。
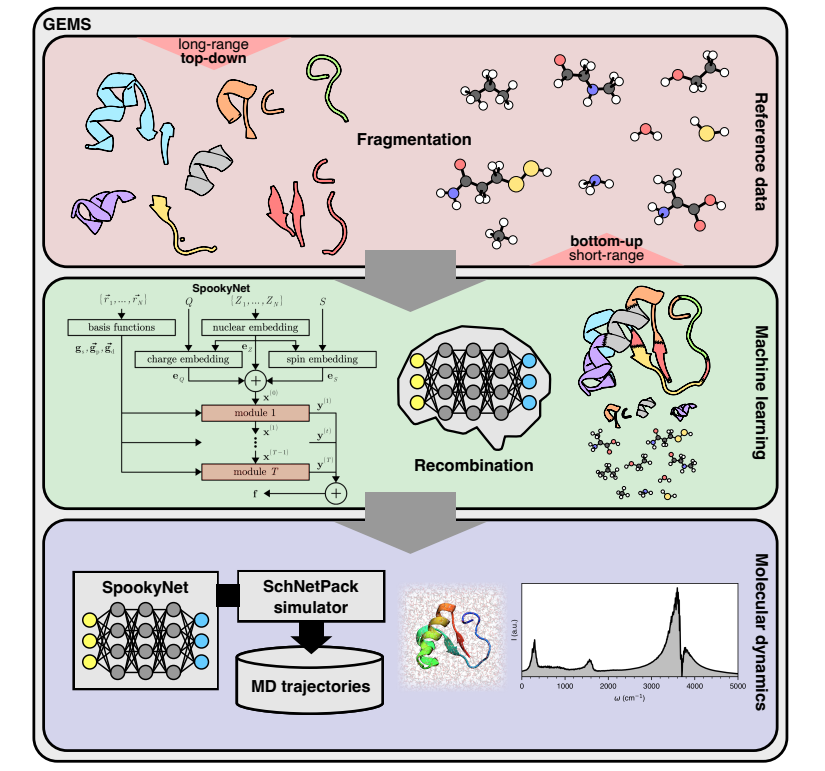
基于分而治之的原则,大型异质系统的 MLFF 在不同大小的分子片段上进行训练,这些分子片段仍然适合电子结构计算。这些碎片并不构成更大系统的分区;相反,它们可以是重叠的部分,甚至只是在结构上与原始系统相关。
在评估 MLFF 时,不会直接使用这些片段,而是仅在训练过程中使用,从而了解较大系统中存在的相关物理化学相互作用。
根据这些片段数据(包括水或溶剂分子),ML 模型推断重组原始系统,并能够预测完整的势能表面(PES),包括与溶剂的相互作用,这使得 GEMS 能够成功解决从头开始质量生物分子模拟的长期挑战。因此,GEMS 指的是使用以这种方式构建的 MLFF 运行分子模拟的一般原理。
虽然 MLFF 可以成功地从小分子中学习局部化学相互作用,但需要足够数量的较大片段来学习推广到更大系统所需的远程效应,并相对于从头开始的基本事实实现高预测精度(能量为 0.450 meV/atom,力为 36.704 meV/Å)。
在这里,科学家依赖于最近提出的 SpookyNet 架构,该架构通过将物理驱动的交互项嵌入到 ML 架构中并从参考数据中学习其参数来显式地模拟色散和静电。
研究人员注意到 SpookyNet 模型并不是第一个明确模拟远程静电的模型,其他模型也遵循类似的方法。此外,原子核之间短程排斥力的经验项提高了模型对强键畸变的稳健性。
SpookyNet 还包括一种描述非局部电荷转移等效应的机制,而其他 MLFF 通常无法做到这一点。当使用适当的参考数据进行训练时,这些组件共同使模型能够推广到更大的分子。
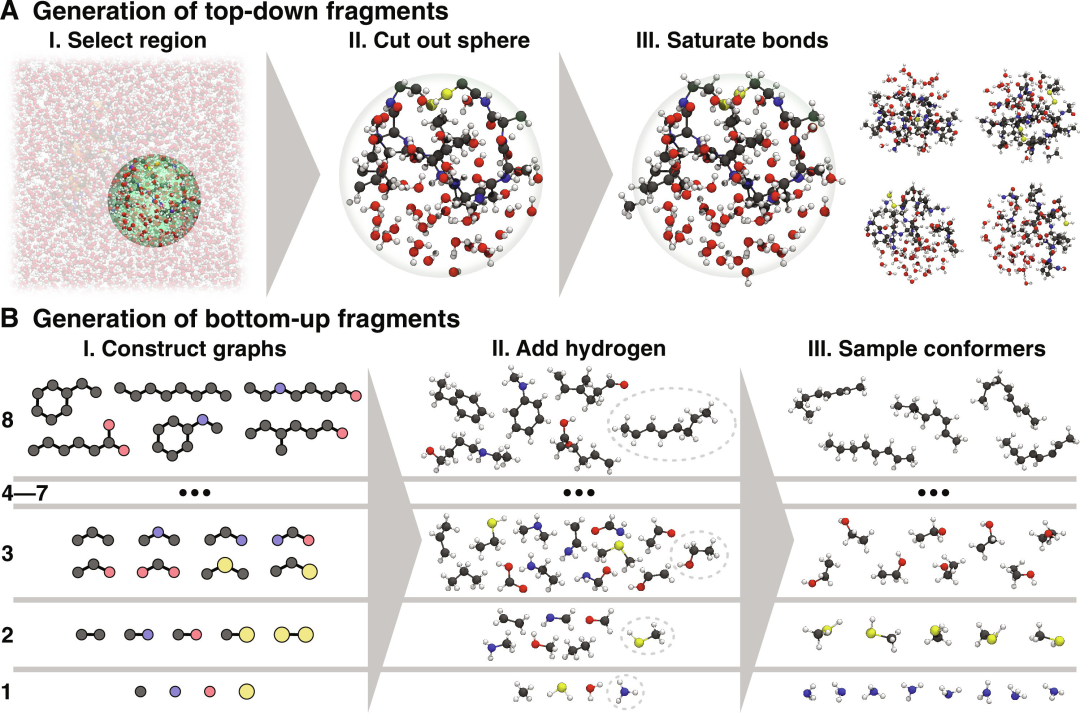
图示:自上而下和自下而上片段的生成。(来源:论文)
至关重要的是,这使得 GEMS 能够解释合作的远程效应,这对于传统的 FF 来说是困难或不可能的。虽然小片段的大量参考数据主要用于学习短程相互作用的稳健「基线」表示,但额外的较大片段允许 GEMS 还捕获长程相互作用以及不同相互作用尺度之间的相互作用。用同样的方式,也可以囊括溶剂效应(通过明确描述与溶剂分子的相互作用)。
研究证明,GEMS 可以学习从此类片段数据中准确地模拟大规模现象,例如协作偏振效应,从而与从头开始的地面事实实现密切一致。
然而,MLFF 的质量和可靠性应该通过其对实验测量的预测来判断,例如,GEMS 能够定量重现有关不同温度下聚丙氨酸系统螺旋稳定性的实验结果,并正确描述溶剂化的 46 个残基蛋白质(crambin)的太赫兹红外(IR)振动光谱。
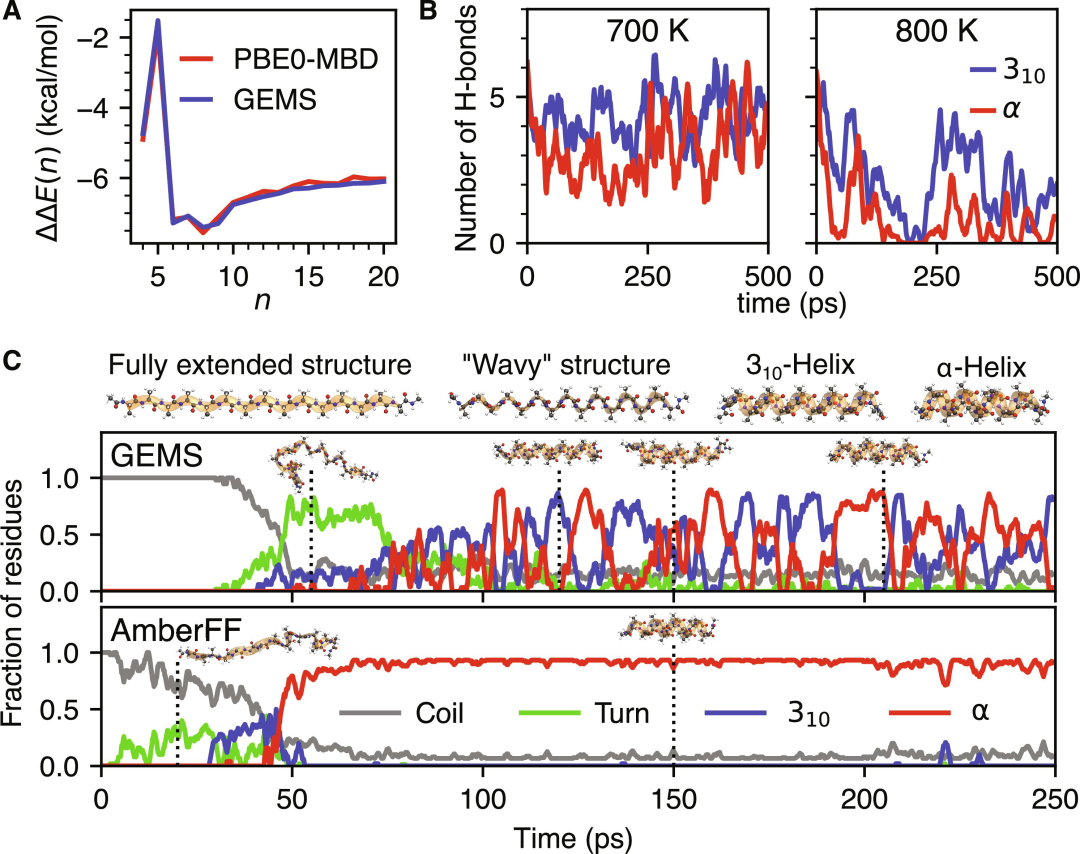
图示:使用 GEMS 精确模拟聚丙氨酸系统。(来源:论文)
使用传统的经验 FF 很难实现这一点,传统的经验 FF 不考虑集体多体相互作用,因此产生最多是定性的大规模振动模式,通常会在 25 至 150 cm−1 光谱区域内出现峰结构的模糊和振幅的夸大。
GEMS 适用于模型肽和含 8205 个明确水分子(>25,000 个原子)的水溶液中的 46 残基蛋白质 Crambin 的 MD 模拟。与传统的 FF(例如 AMBER99SB-ILDN)相比,GEMS 更接近根据密度泛函理论计算的能量和力。
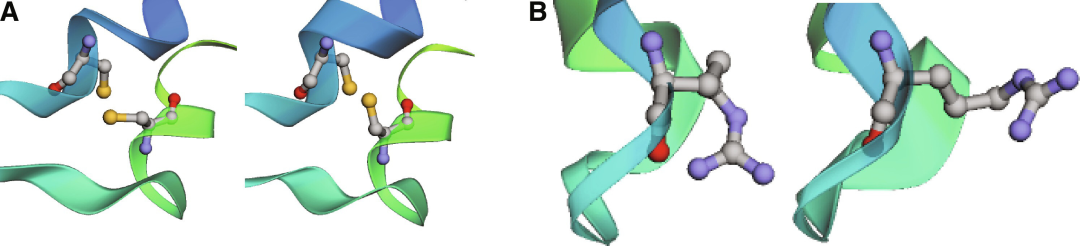
图示:Cambin 中的半胱氨酸/精氨酸残基。(来源:论文)
研究结果揭示了聚丙氨酸肽折叠途径中以前未知的中间体以及 α-螺旋和 310-螺旋之间的动态平衡。
在溶剂化 Crambin 的模拟中,GEMS 表明蛋白质运动在性质上有所不同,与传统 FF 的计算相比,PES 更平滑,振动更柔和,显示出对比的短时标和长时标动态。
低频振动模式很大程度上决定了蛋白质的自由能;该团队的结果表明,为了充分理解生物分子的动态过程,可能需要从头开始进行精确地计算模拟。
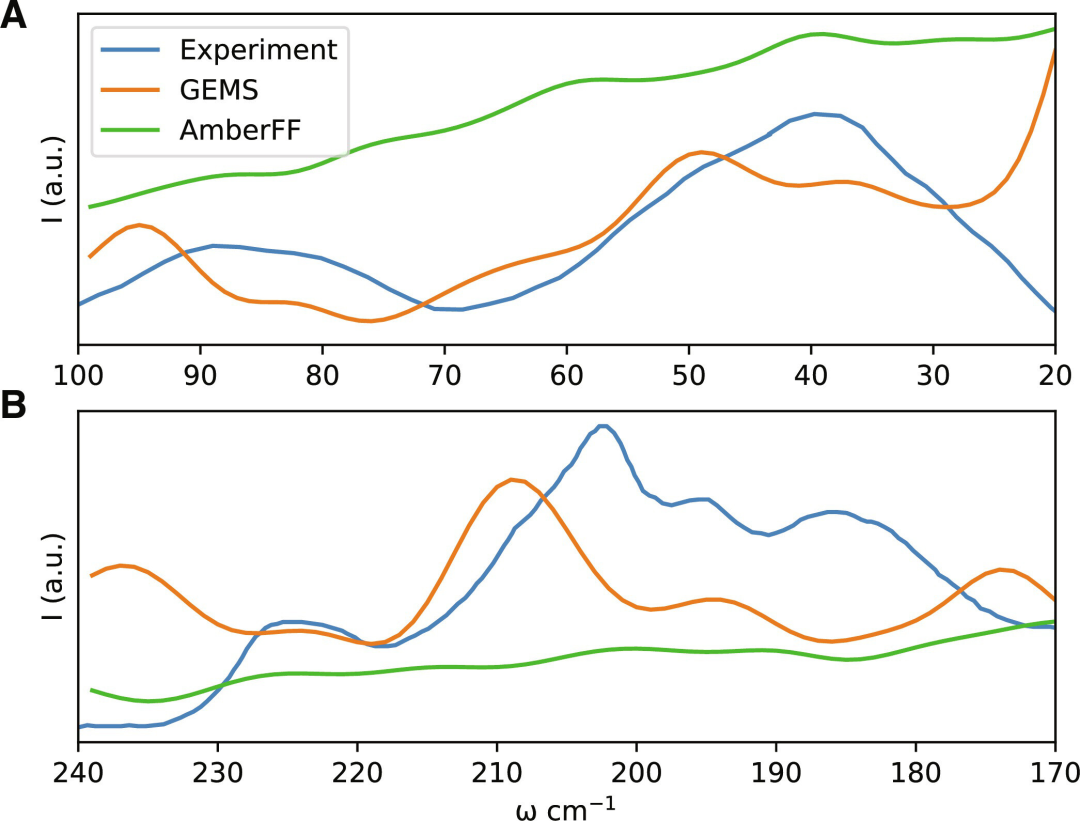
图示:Crambin 在太赫兹时间尺度上的红外光谱。(来源:论文)
结语
GEMS 的未来工作可能包括将其扩展到更大系统和更长时间尺度的模拟,以及可能的扩展包括纳入核量子效应,这些都可能为研究大分子系统的动态提供新的视角。
虽然 GEMS 在计算效率上优于从头算计算,但仍低于传统 FF。此外,GEMS 在评估时通常需要更多的内存,这限制了可模拟的最大系统大小。尽管如此,GEMS 仍然能够在保持从头算精度的同时模拟数千原子系统的几纳秒动态。
使用精确的 MLFF 的另一个优势在于能够获得任意导数,包括化学衍生物。这可能使得针对局部突变优化可观测量成为可能,从而在药物设计和蛋白质工程中发挥重要作用。
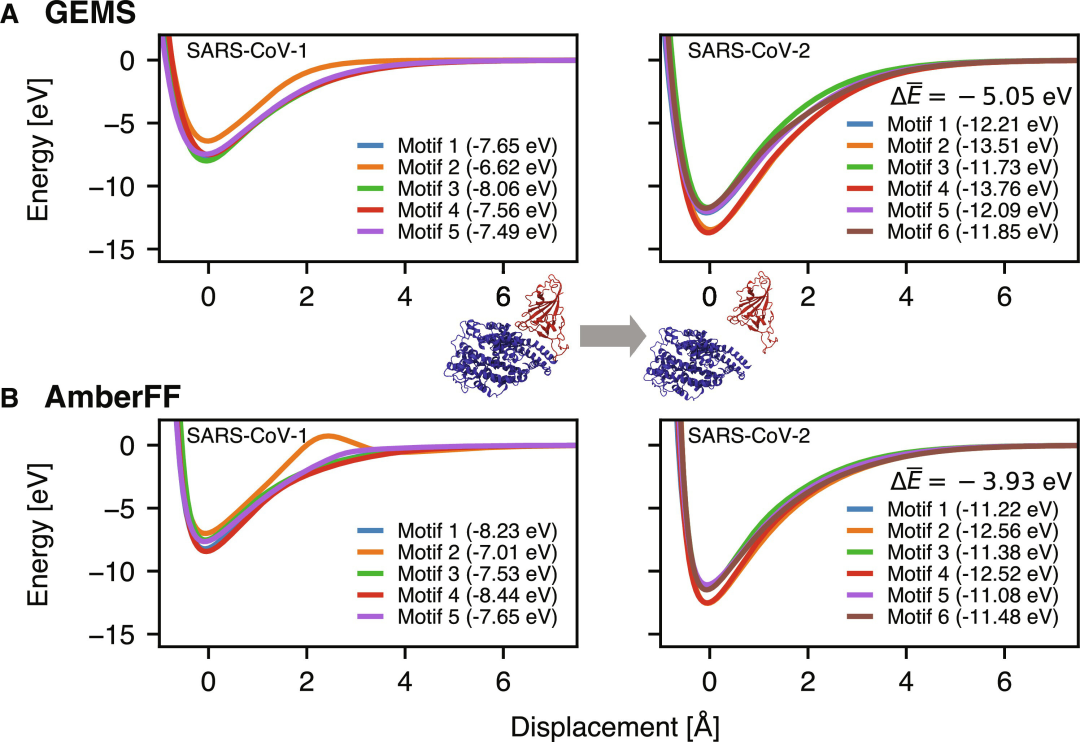
图示:实现精确的量子力学蛋白质-蛋白质相互作用:ACE2(蓝色)和 SARS-CoV 刺突蛋白 RBD(红色)的气相结合曲线。(来源:论文)
GEMS在模拟蛋白质-蛋白质相互作用方面展现出应用潜力,例如在模拟 ACE2 与 SARS-CoV-1 和 SARS-CoV-2 变体的 RBD 结合时,GEMS 提供了更强的结合能预测,这可能对理解病毒如何与宿主细胞相互作用具有重要意义。
虽然目前GEMS使用的片段是特定于系统的,但未来的发展可能会使其能够覆盖更广泛的系统范围,从而实现具有化学可转移性和尺寸可扩展性的「通用」MLFF,这将进一步推动 GEMS 在多个领域的应用。
论文链接:https://www.science.org/doi/10.1126/sciadv.adn4397
上一篇:电脑上免费听音乐的软件
产品推荐
-

售后无忧
立即购买>- DAEMON Tools Lite 10【序列号终身授权 + 中文版 + Win】
-
¥150.00
office旗舰店
-

售后无忧
立即购买>- DAEMON Tools Ultra 5【序列号终身授权 + 中文版 + Win】
-
¥198.00
office旗舰店
-

售后无忧
立即购买>- DAEMON Tools Pro 8【序列号终身授权 + 中文版 + Win】
-
¥189.00
office旗舰店
-

售后无忧
立即购买>- CorelDRAW X8 简体中文【标准版 + Win】
-
¥1788.00
office旗舰店
-
 正版软件
正版软件
- okex购买狗狗币单笔下限数
- OKEx购买狗狗币的单笔下限为10美元。单笔下限旨在防止市场操纵和投机,并提高交易效率。若要绕过该限制,可合并多个订单或在其他交易所交易。
- 刚刚 0
-
 正版软件
正版软件
- okex交易所是什么
- OKEx是一家成立于2017年的全球数字资产交易所,总部位于塞舌尔,是世界上交易量最大的加密货币交易所之一。它的特点包括高流动性,广泛的交易对,先进的交易工具,以及安全性和合规性。OKEx的优势包括:广泛的数字资产选择,出色的流动性,高级交易工具,严格的安全和合规措施,以及全球影响力和支持。
- 10分钟前 0
-
 正版软件
正版软件
- dash币有希望吗
- Dash币的前景不确定,既有增长机会也有挑战。其优势包括注重隐私、快速交易和去中心化治理。挑战包括竞争激烈、监管不确定性和技术限制。潜在的增长机会在于隐私需求的增长、大额交易的采用和去中心化治理的演变。最终,Dash币的成功取决于其团队能否克服挑战并利用增长机会。
- 25分钟前 0
-
 正版软件
正版软件
- 2024 年2月NFT行业动态:加密货币飙升,NFT市场调整
- 数据来源:NFT研究页面-FootprintAnalytics2024年2月,加密货币与NFT市场显现出了复杂性。该月,NFT领域的交易量达到12亿美元,环比下降了3.7%。值得关注的是,包括Azuki、MAYC和BAYC等在内的知名NFT系列,在以太坊、Polygon、BNB链、Cronos、Optimism和Sui等公链上,均出现了显著的交易量下滑,降幅高达32.1%。此外,GasHero游戏NFT表现不佳,波及到了Polygon和Mooar等平台。与此同时,新的叙事如ERC404和DN404在此期间
- 40分钟前 关键词 字数 0
-
 正版软件
正版软件
- 梅赛德斯-AMG ONE超跑:五年磨一剑 限量275台震撼上市
- 7月4日消息,近日,来自F1车手博塔斯的个人社交媒体上晒出了他新获得的一台AMGONE跑车。这款梅赛德斯-AMGONE公路超跑备受瞩目,搭载一级方程式赛车发动机,其吸引力不言而喻。不仅如此,这款超跑还赢得了许多名人的青睐,其中包括现今的两位F1车手汉密尔顿和博塔斯。据了解,AMGONE于2017年9月在法兰克福车展上首次亮相,经历了漫长的五年周期,直到2022年8月正式开售,2023年1月开始正式生产,方才交付给买家。博塔斯在晒出新车照片的同时,只留下了“值得等待(Worththewait.)”的字样,显
- 55分钟前 0
最新发布
-
 1
1
- 阿里追捧的中台,“热度”退了?
- 1881天前
-
 2
2
- Overture设置踏板标记的方法
- 1718天前
-
 3
3
- 思杰马克丁取得CleanMyMac中国区独家发行授权
- 1708天前
-
 4
4
- IBM:20万台Mac让公司职工在工作中更快乐 更多产
- 1906天前
-
 5
5
- 报道称微软一直在悄然游说反对“维修权”立法!
- 1872天前
-
 6
6
- 美国怀疑华为窃取商业机密 华为:身正不怕影子斜
- 1868天前
-
 7
7
- 三星被曝正与联发科接洽 A系列手机有望搭载其5G芯片
- 1883天前
-
 8
8
- 环球墨非完成千万级融资 联合企业集团投资
- 1904天前
-
 9
9
相关推荐
热门关注
-

- Xshell 6 简体中文
- ¥899.00-¥1149.00
-

- DaVinci Resolve Studio 16 简体中文
- ¥2550.00-¥2550.00
-

- Camtasia 2019 简体中文
- ¥689.00-¥689.00
-

- Luminar 3 简体中文
- ¥288.00-¥288.00
-

- Apowersoft 录屏王 简体中文
- ¥129.00-¥339.00