上科大团队开发了一种基于Transformer编码器和指纹图谱的药物-靶标亲和力预测方法
 发布于2024-11-21 阅读(0)
发布于2024-11-21 阅读(0)
扫一扫,手机访问

编辑 | 萝卜皮
药物发现的关键之一是预测药物与靶标之间的结合亲和力,但目前现有方法的准确性还有待提高。此外,大多数深度学习方法只关注非共价结合分子系统的预测,而忽略了共价结合在药物开发领域的重要性。因此,研究人员需要进一步改进现有的方法,以提高结合亲和力的预测准确性,并且考虑到共价结合的情况。这将有助于加速药物发现过程,并为药物开发提供更多有价值的信息。
上海科技大学的研究团队提出了一种新的基于注意力的模型,名为TEFDTA(Transformer Encoder and Fingerprint combined Prediction method for Drug-Target Affinity)。该模型旨在预测药物与靶标之间的结合亲和力,不论是键合还是非键合相互作用。通过利用注意力机制,该模型能够准确地预测药物-靶标的相互作用,并为药物研发和靶向治疗提供重要指导。这一研究成果对于加速药物发现和开发具有重要意义,并为未来的药物研究提供了新的思路和方法。
为了应对这一复杂问题,研究人员采用了不同的表示方法来研究蛋白质和药物分子。他们通过使用非键合蛋白质-配体相互作用数据集来训练模型,从而构建了初始框架。
该团队为广泛使用的数据集Davis提供了手动校正的Davis数据库,同时也对该模型在CovalentInDB数据库中的较小共价相互作用数据集上进行了微调,以优化性能。
结果表明,与单独使用 BindingDB 数据相比有了显著改进,预测非共价结合亲和力平均提高了 7.6%,预测共价结合亲和力平均提高了 62.9%。
该研究以「TEFDTA: a transformer encoder and fingerprint representation combined prediction method for bonded and non-bonded drug–target affinities」为题,于 2023 年 12 月 23 日发布在《Bioinformatics》。

在药物研发领域,预测药物与靶点相互作用/亲和力(DTI/DTA)是不可或缺的组成部分。
在早期阶段,研究人员通过实验确定这些相互作用,这既耗时又昂贵。随着计算机技术的进步,研究人员开始利用计算机来预测药物与靶点的相互作用,并使用对接程序(例如 GLIDE、Molegro Virtual Docker)模拟药物与靶点的结合姿势。但这种对接方法也有相应的局限性,即对接过程也需要较长的计算时间,并且需要蛋白质的三维结构。
随着机器学习和深度学习的发展,研究人员尝试将这些领域纳入DTI。目前,基于深度学习的方法已得到广泛应用。这些方法的优点是能够自动提取特征。然而,初始输入数据,特别是蛋白质和小分子的数据描述,显著影响模型的性能。
在最新的研究中,上海科技大学的研究团队提出了一种用于预测药物-蛋白质相互作用中的共价(键合)和非共价(非键合)结合亲和力的新模型,称为指纹编码器 DTA (TEFDTA)。
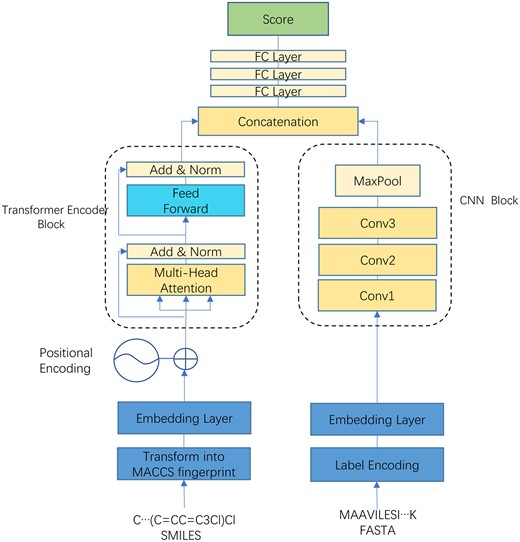
TEFDTA 从两个现有模型 DeepDTA 和 TransformerCPI 中汲取灵感。DeepDTA 提供了一种使用 1D-CNN(一维卷积神经网络)从序列中提取特征的方法。该模型侧重于从序列信息中提取局部模式特征,以方便特征提取。虽然循环神经网络(RNN)也可以处理一维输入并执行特征提取,但它们存在某些局限性。
另一方面,CNN 在有效捕获全局特征方面存在局限性。RNN 尽管能够通过网络传播处理整个序列,但会遇到随着时间的推移而忘记信息的问题。TransformerCPI 证明 Transformer 可以有效解决 CNN 和 RNN 模型中存在的问题。Transformer 构建在编码器和解码器之上。
鉴于此,该团队利用 Transformer 作为特征提取器来提取复杂的分子序列。值得注意的是,单个编码器足以完成此任务,因为由于潜在的收敛困难,更复杂的模型将需要更长的训练时间,而不必增强信息提取。
为了评估 TEFDTA 的性能,研究人员在 Davis、KIBA 和 BindingDB 数据集上进行了实验,并将结果与其他结合亲和力预测模型(即 DeepDTA 和 DeepCDA)进行了比较。
结果证实了 TEFDTA 在结合亲和力预测方面的性能。此外,通过对数据库 CovalentInDB 中键合蛋白-配体相互作用的数据集进行微调,进一步优化了该模型。共价结合数据根据常见弹头进行分类,并对每个弹头类别进行单独微调。结果表明,微调过程显著提高了模型对共价结合亲和力的预测准确性,强调了专门训练的重要性。
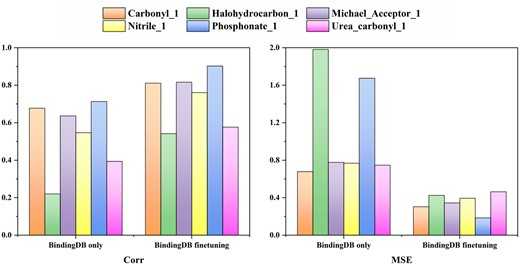
图示:六种常见弹头微调前后的共价结合亲和力预测比较。(来源:论文)
此外,该团队还进行了预测针对 EGFR 的药物分子的结合亲和力的案例研究。结果表明,虽然该模型可能无法精确预测具有相同主链结构但取代基不同的分子的确切结合亲和力值,但它能够捕获分子上不同取代基引入的亲和力方差的趋势。这表明该模型对局部结构变化的潜在敏感性及其近似结合亲和力强度的能力,需要用更大的数据集进行确认以进行进一步的评估或训练。
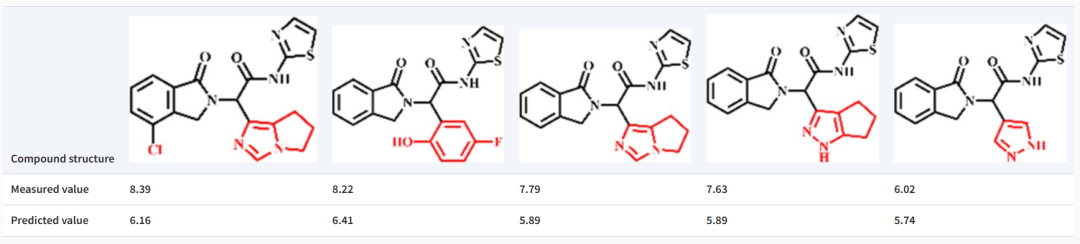
图示:TEFDTA 对区分由微小结构差异引起的结合亲和力 (pKd) 差异的敏感性的结果。(来源:论文)
总之,TEFDTA 结合了指纹变换和 Transformer 编码器模块,为准确预测药物-靶标相互作用提供了一种改进的方法。
同样该模型也存在一些局限性。虽然该模型可以成功捕获分子序列的微小变化对亲和力的影响,但它对蛋白质片段的突变(包括单个或几个氨基酸变化)并不敏感。对于虚拟筛选任务,当突变发生时检测亲和力的可观察变化非常有价值。
直接从蛋白质的 FASTA 序列中提取特征很难实现这一目标,因为单个氨基酸突变在整个蛋白质的嵌入中是难以察觉的。然而,随着大型语言模型的出现,通过在大量蛋白质序列上预训练这些模型,通过无监督学习来提取蛋白质的表示已经成为可能。通过使用下游任务(例如具有突变的数据)对模型进行微调,模型变得对关键氨基酸敏感。
未来,该团队还将尝试使用大型语言模型来提取蛋白质表示。此外,目前对共价结合亲和力的预测需要先验了解配体和靶标对的共价键类型,这可能限制了广泛和正确的应用,特别是对于非化学家来说不友好。这些方向值得在未来的研究工作中进一步探索。
论文链接:https://academic.oup.com/bioinformatics/article/40/1/btad778/7492659
产品推荐
-

售后无忧
立即购买>- DAEMON Tools Lite 10【序列号终身授权 + 中文版 + Win】
-
¥150.00
office旗舰店
-

售后无忧
立即购买>- DAEMON Tools Ultra 5【序列号终身授权 + 中文版 + Win】
-
¥198.00
office旗舰店
-

售后无忧
立即购买>- DAEMON Tools Pro 8【序列号终身授权 + 中文版 + Win】
-
¥189.00
office旗舰店
-

售后无忧
立即购买>- CorelDRAW X8 简体中文【标准版 + Win】
-
¥1788.00
office旗舰店
-
 正版软件
正版软件
- 福特Mustang Dark Horse即将在国内上市——力量与美感的完美融合的倒计时
- 福特中国今日揭开了备受期待的全新MustangDarkHorse车型的面纱,宣布该款美式肌肉车将于2月19日正式上市,并计划在今年第二季度开始交付给消费者。这款车型备受期待,翘首以盼的消费者将很快能够亲眼目睹并驾驭它。福特中国为车迷们带来了一款更加神秘、更加具有冲击力的Mustang车型,相信它将成为市场瞩目的焦点。新款MustangDarkHorse保留了Mustang系列一贯的坚韧风格,车身线条刚毅且充满力量感。前脸部分,该车采用了熏黑处理的格栅,搭配大尺寸散热开口的发动机舱盖,展现出强烈的运动气息。
- 13分钟前 福特 0
-
 正版软件
正版软件
- 2024年ICLR国际顶级会议揭晓,蚂蚁集团获选11篇论文
- 最近,ICLR2024,人工智能顶级会议,公布了录取结果。蚂蚁集团在这次会议中有11篇论文被接收,其中1篇被评为口头报告,3篇被选为焦点报告,另外7篇则为海报展示。蚂蚁集团在人工智能学术界的进展备受瞩目。(图:蚂蚁集团的《长视频中的多粒度噪声关联学习》被收录为Oral论文)今年ICLR组委会收到了7262篇论文投稿,录用率约为31%。根据录用结果,其中1.2%的论文被录用为Oral论文,这些作者将获得10分钟的口头演讲机会。另外5%的论文被录用为Spotlight论文,这些作者将有4分钟的聚光灯展示时间。
- 18分钟前 入门 ICLR 噪声关联 多模态技术 关联学习 0
-
 正版软件
正版软件
- 中国团队发现室温超导续集?最新论文证实LK-99可能出现迈斯纳效应,刚刚发布
- 一篇新的室温超导论文再次引起互联网上的轻微波动。最新论文中再次证明了室温下铜可能替代铅磷灰石(LK-99)中的迈斯纳效应存在。论文链接:https://arxiv.org/pdf/2401.00999.pdf,供大家参考。在室温下,用铜取代的铅磷灰石在25Oe的磁场下观察到抗磁性直流磁化,在零场冷却和场冷却测量之间存在明显的分歧,在200Oe下变为顺磁性。在冷却过程中发现了玻璃记忆效应。超导体的典型磁滞回线在250K以下被检测到,同时磁场的前后扫描不对称。我们的实验表明,在室温下,这种材料可能存在迈斯纳效
- 28分钟前 模型 AI 0
-
 正版软件
正版软件
- 中国公司统治苹果供应链:Vision Pro席卷市场
- 6月16日消息,苹果在今天的WWDC23开发者大会上正式发布了其首款增强现实产品VisionPro。该产品的主要供应商超过一半来自中国,其中包括立讯精密、高伟电子、德赛电池等中国厂商。根据发布的物料清单显示,苹果VisionPro的供应商数量中超过一半来自中国,其中包括7家大陆地区厂商和11家台湾地区厂商。大陆厂商中除了立讯精密外,还有高伟电子、德赛电池、歌尔股份、长盈精密、领益制造、兆威机电。台湾地区厂商则包括台积电、大立光、业成集团、日月光、和硕、台郡、臻鼎、欣兴、景硕、南电、采钰。据小编了解,苹果V
- 41分钟前 0
-
 正版软件
正版软件
- 年轻人首选的先锋潮机,华为 nova 12 系列超强综合体验正式上市!
- 2023年12月26日,问界M9及华为冬季全场景发布会上,全新的华为nova12系列正式亮相,这一次,nova12系列发挥“时尚先锋全能人像”定位优势,选用极为瞩目的蓝色作为主打色,打造吸睛的12号色,影像部分搭载了第二代物理可变光圈镜头,搭配全新鸿蒙智慧通信和HarmonyOS4等便捷体验,让年轻人在2024年新年拥有更多购机选择。今日,华为nova12及nova12Pro已正式迎来首销,下周的1月12日,华为nova12Ultra也将正式开售。“蓝”不住的实力,“蓝”不住的潮流,如果你需要一款能够时刻
- 53分钟前 Nova nova12 0
最新发布
-
 1
1
- 阿里追捧的中台,“热度”退了?
- 1807天前
-
 2
2
- Overture设置踏板标记的方法
- 1644天前
-
 3
3
- 思杰马克丁取得CleanMyMac中国区独家发行授权
- 1634天前
-
 4
4
- IBM:20万台Mac让公司职工在工作中更快乐 更多产
- 1832天前
-
 5
5
- 报道称微软一直在悄然游说反对“维修权”立法!
- 1798天前
-
 6
6
- 美国怀疑华为窃取商业机密 华为:身正不怕影子斜
- 1794天前
-
 7
7
- 三星被曝正与联发科接洽 A系列手机有望搭载其5G芯片
- 1809天前
-
 8
8
- 环球墨非完成千万级融资 联合企业集团投资
- 1830天前
-
 9
9
相关推荐
热门关注
-

- Xshell 6 简体中文
- ¥899.00-¥1149.00
-

- DaVinci Resolve Studio 16 简体中文
- ¥2550.00-¥2550.00
-

- Camtasia 2019 简体中文
- ¥689.00-¥689.00
-

- Luminar 3 简体中文
- ¥288.00-¥288.00
-

- Apowersoft 录屏王 简体中文
- ¥129.00-¥339.00