纯AI开发新药在Nature杂志发表,提高研发效率3倍,临床试验表现出色
 发布于2024-12-18 阅读(0)
发布于2024-12-18 阅读(0)
扫一扫,手机访问
完全由AI研发的药物马上将要上市了?!
由AI制药公司Insilico Medicine开发的治疗肺部纤维化的药物TNIK已经进入二期临床试验。
研究团队的在Nature子刊上发表了最新的研究成果。

论文地址:https://www.nature.com/articles/s41587-024-02143-0#Sec14
该论文详细描述了一个名为INS018_055的药物研发过程。这种药物是Insilico Medicine利用他们开发的AI平台PHARMA.AI发现的潜在治疗方案,用于对抗一种罕见但致命的肺部纤维化疾病。
这个研发过程开创了生成式人工智能在药物发现领域的先例。
该药物是世界首个利用生物AI发现靶点并确定优先次序,同时利用化学AI生成分子的药物。

而发现这种药物的平台Pharma.AI,包含了多个数百万个数据样本上训练出来的多个AI模型,用于完成一系列任务。

其中一个PandaOmics 可以快速识别对疾病疗效起重要作用的靶点,并确定其优先次序。
另一个工具是Chemistry42引擎,它利用深度学习技术,可以快速设计出针对PandaOmics所识别的蛋白质的新的潜在药物化合物。
传统的药物开发通常是一条「非常漫长但也非常危险的道路」,需要数十年的临床前细胞、组织、动物模型和人体临床试验,耗资数十亿美元,失败率超过90%。
据而根据Insilico估算,如果采用传统的药物发现方法,和INS018_055相似的新药需要花费4亿多美元,时间可能长达6年。
但利用生成式AI,该公司仅用两年半的时间就完成了临床试验的第一阶段,而成本仅为原来的1/3。
AI药物平台开发新药,效率提高3倍
特发性肺纤维化(IPF)是一种侵袭性间质性肺病,死亡率很高。
而目前和IPF的潜在药物靶点未能转化为临床层面的有效疗法。
研究人员采用预测性人工智能(AI)方法,将TRAF2-与NCK相互作用激酶(TNIK)确定为抗纤维化靶点。
再进一步利用人工智能驱动的方法,生成了一种小分子TNIK抑制剂 INS018_055,它通过口服、吸入或局部给药,在体内不同器官中表现出理想的类药物特性和抗纤维化活性。
而且INS018_055除具有抗纤维化特性外,还具有抗炎作用,这已在多项体内研究中得到验证。
一项随机、双盲、安慰剂对照的 I 期临床试验(NCT05154240)验证了 INS018_055的安全性、耐受性和药代动力学,78名健康人参加了该试验。
在中国进行的另一项 I 期临床试验(CTR20221542)也证明了该药物具有可比的安全性和药代动力学特征。
从目标发现到临床前候选药物提名,整个流程在大约18个月内完成,展示了生成式人工智能驱动药物发现的能力。
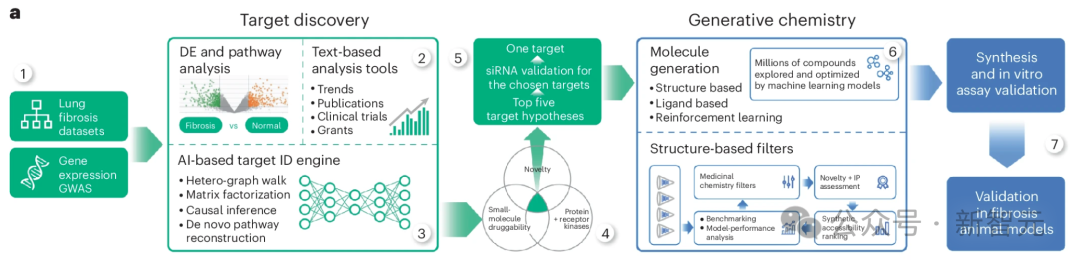
治疗靶点的确定是包括纤维化在内的所有疾病的药物研发过程中至关重要的一步。在药物开发的早期阶段选择错误的靶点可能会导致药物发现项目成本高昂,并往往在多年后的二期试验中以失败告终。
因此,大型制药公司可能会表现出规避风险的态度,从而降低对有潜在价值的靶点和治疗策略进行投资的意愿。
尽管开发精确的数据驱动药物靶点发现方法对临床试验的成功有着至关重要的影响,但这项任务仍存在公认的局限性,如数据复杂性和批量效应。
最近,人工智能驱动的方法已在胚胎-胎儿转化7 和肌肉衰老方面证明了发现候选靶点的有效性。
应用于多组学数据的高级通路分析和人工智能算法即使在先前证据稀少的情况下也能发现靶点和生物标记物。
IPF是一种逐渐进展且通常致死的疾病,组织学特征为成纤维细胞增殖和大量细胞外基质沉积。
IPF 最常见于60岁以上的患者,据报道,其在美国的估计发病率为每10万人年6.8例。
由于发病率不断上升,治疗IPF患者的费用给医疗系统造成了沉重负担,并成为全球日益严重的公共卫生问题。
在未经治疗的患者中,IPF 的临床病程极不稳定,中位生存期为2到3年。
研发流程
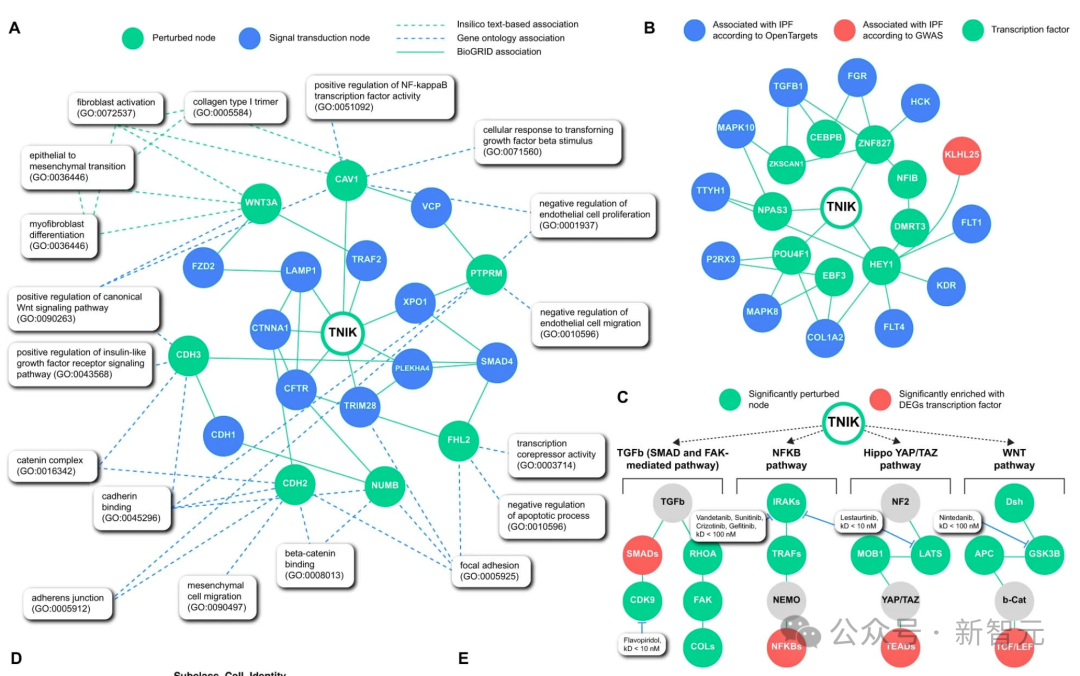
研究人员的人工智能生成平台发现TNIK抑制是一种有效的抗纤维化策略,并协助开发了一种高度特异性的TNIK抑制剂INS018_055。
在这里,使用PandaOmics 这一商用靶点发现平台,利用包括生成预训练转换器在内的多种人工智能引擎,进行纤维化的靶点识别。
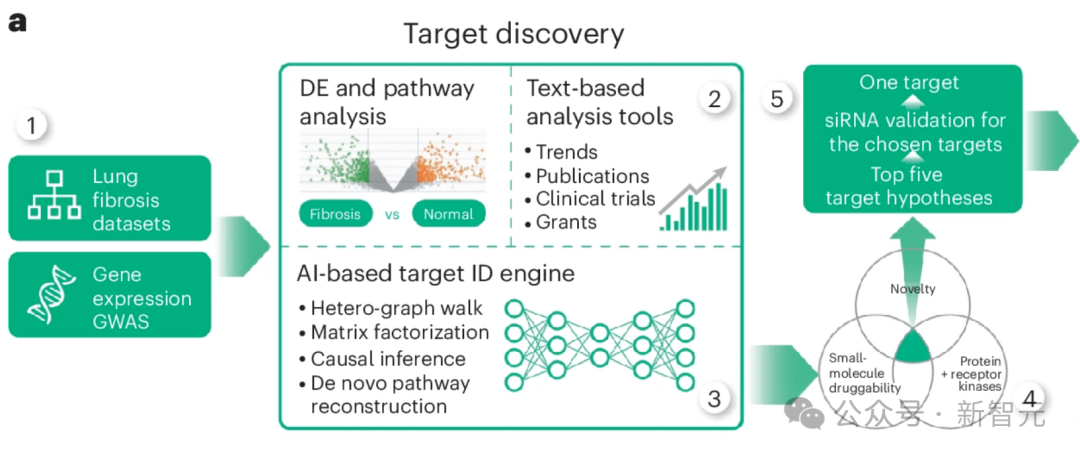
研究人员将从 IPF 患者组织样本中提取的多组学数据集列表作为计算管道的基础(图 1a,步骤 1)。
这个流程结合了几种不同但互补的计算方法,所有方法的输出结果(按最有希望的靶点排序的基因列表)相似,但输入数据不同。
除多组学数据集外,研究人员还采用了依赖生物网络分析和科学文献文本数据的方法(图 1a,步骤 2),并对节点度偏差进行了控制。
这些数据类型有助于将从omics中推断出的目标假设与先前的证据(包括临床试验、出版物和拨款申请)结合起来。
文本数据具有时间性,使 PandaOmics 不仅能捕捉趋势,甚至还能预测未来(图 1a,步骤 3)。
与 Open Targets43 或 NewDrugTargets44 等其他靶点发现方法不同,研究人员的方法是为搜索多种疾病和老龄化的靶点而设计并经过验证的。
因此,研究人员开发了一种时间机器方法,利用某个时间点之前发布的数据训练计算模型,并通过模型预测该时间点之后制药业关注的靶点的能力来验证模型输出结果。
研究人员应用了几种不同的人工智能驱动的靶点发现理念,包括异质图上的随机漫步和负矩阵因式分解(补充视频 1)。
研究人员的平台在综合应用分数构成和过滤器(包括蛋白质家族等疾病诊断特性、小分子或治疗性抗体的可及性、新颖性和晶体结构可用性等)后生成了一份目标排序列表。
为了生成纤维化的目标假设,研究人员对纤维化相关数据集采用了 「蛋白质和受体激酶 」方案。
它包括反映疾病机制网络成分的分数,以及基于转录因子富集的因果推断(补充信息 1)。在下游处理步骤中启用了蛋白质类别(仅限蛋白质和受体激酶)、新颖性和小分子药物筛选器(图 1a,步骤 4)。
使用 「蛋白和受体激酶 」方法,TNIK被确定为五大候选靶点中的第一位,其网络邻域、因果推断、通路、相互作用组群落、表达、异质图步行和矩阵因式分解得分值相对较高(图 1a,步骤 5,图 1b)。
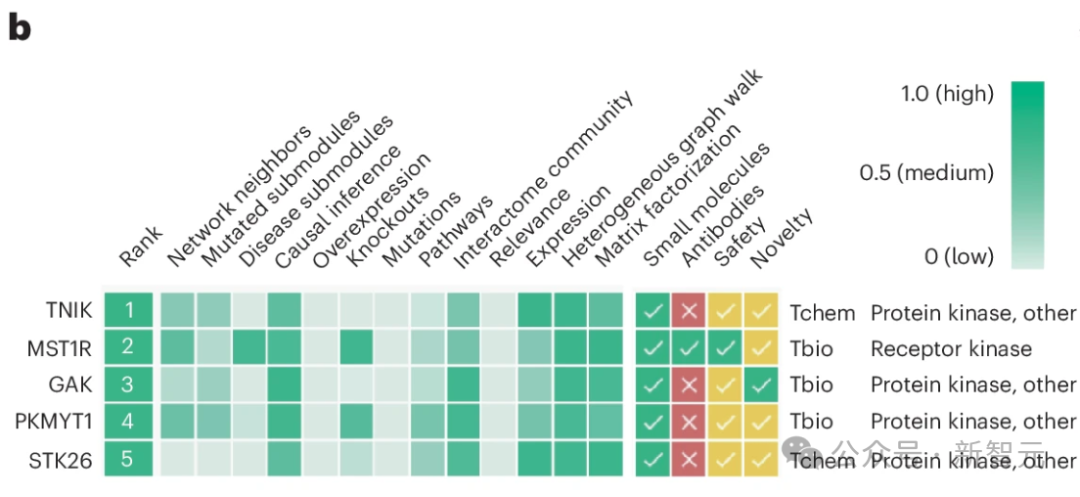
TNIK 与纤维化驱动通路相关,包括 WNT45,46、TGF-β46,47、Hippo(是相关蛋白(YAP)-具有 PDZ 结合基调(TAZ)的转录辅激活因子)、JNK和核因子(NF)-κB信号转导。
不过,TNIK尚未作为IPF的治疗靶点进行研究,因此被人工智能算法选中。
虽然酪氨酸激酶抑制剂如宁替尼、伊马替尼和尼洛替尼已在 IPF 中进行了测试,但丝氨酸-苏氨酸激酶(如 TNIK)在 IPF中的作用在很大程度上仍未得到探讨(图 1c)。
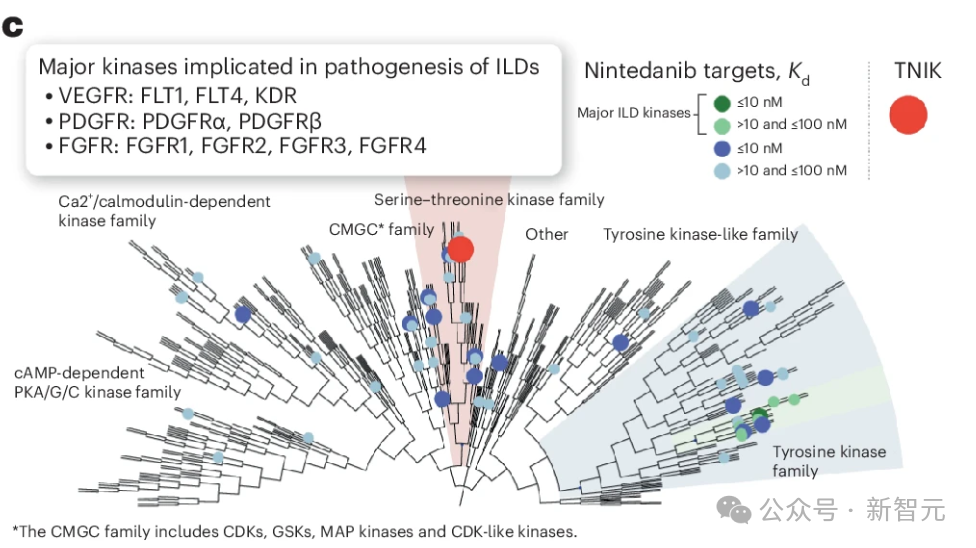
由于纤维化可影响任何器官,并且是工业化国家高达 45% 的死亡原因51,研究人员假设抑制 TNIK 可为多种可能与纤维化有关的病理提供治疗益处。
与这一假设一致的是,TNIK 被独立预测为与多种衰老特征相关的疾病靶点15,并且是衰老和/或高脂饮食喂养小鼠的脂质代谢调节剂52。这些发现支持了研究人员人工智能驱动管道的假设。
接下来,研究人员对 PandaOmics 分数进行了透明度分析。
交互组群落透明度显示,TNIK抑制与多个已知对纤维化进展很重要的生物过程有关,如局灶粘附信号转导、肌成纤维细胞分化和间质细胞迁移(扩展数据图2a)。
因果推理透明度显示,TNIK 与 TGFB1、FGR、FLT1、KDR 等 IPF 相关基因密切相关(下图 2b)。
最后,利用人工智能驱动的新通路重建工具,研究人员发现 TNIK 激活了以前描述的与 IPF 相关的通路。这些基因集受到下游转录因子的调控,包括 TCF-LEF、SMAD、NF-κB 和 TEAD 家族(下图 2c)。
研究人员合成了这种化合物,并在多种鼠类和大鼠纤维化模型中证明了它的选择性抗纤维化活性。
研究人员报告的I期临床试验数据强调了研究人员的小分子抑制剂的安全性和耐受性。从目标发现到临床前候选化合物提名,这一综合方法在大约18个月的时间内完成,展示了研究人员的生成式人工智能驱动药物发现管道的能力。
人工智能设计的 TNIK 抑制剂
为了鉴定TNIK抑制剂,研究人员利用了现有的TNIK激酶结构域晶体结构56 和 Chemistry42 基于结构的药物设计 AI 工作流程(图 1a,步骤 6)。
由于使用ATP竞争者是靶向激酶的成熟策略 ,研究人员选择 ATP 结合位点作为化合物生成的口袋。
人工智能驱动的平台被配置为生成能与 TNIK 铰链区的 Cys108-NH 形成氢键的小分子结构。
除了活性位点外,瞄准保留较少的邻近异生口袋(如靠近守门残基的疏水后腔)也能使先导化合物获得更好的选择性。
因此,研究人员采用了一个额外的疏水药理点,优先考虑具有疏水功能的结构,以深入占据由 Met105、Leu73、Leu103、Ala52 和 Val104 形成的后腔。
根据合成的易得性、新颖性和药物化学特性筛选出的化合物被合成出来,并使用放射性酶测定法进行测试(图 1a,步骤 7)。
第一轮筛选发现了一系列具有纳摩尔级结合亲和力的TNIK抑制剂。
然而,主要先导化合物的体外吸收、分布、代谢和排泄分析表明,它们在人和小鼠肝脏微粒体中的清除率很高,细胞色素 p450(CYP)抑制作用的半最大抑制浓度(IC50)小于 10 µM,动力学溶解度小于 2 µM。
先导物优化阶段优先考虑改善这类化合物的吸收、分布、代谢和排泄情况,最终产生了INS018_055(WO2022179528A1)。
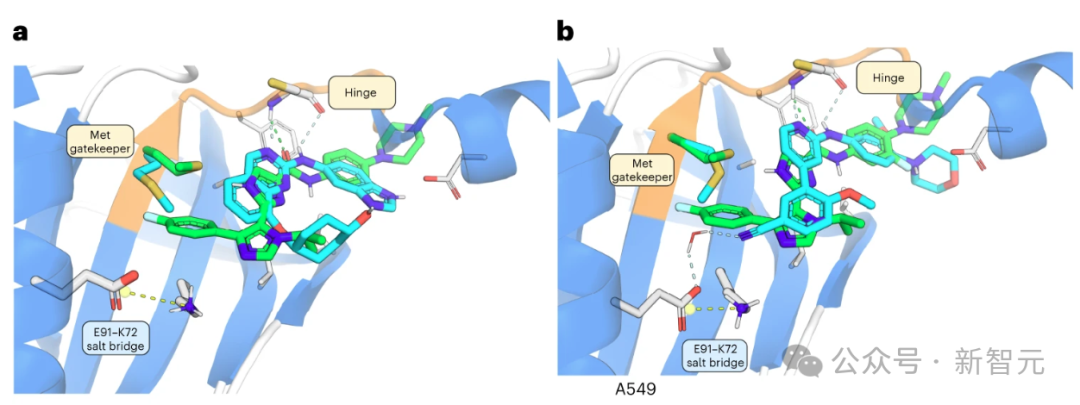
INS018_055 的羧基氧与铰链区内的 Cys108-NH 形成氢桥(图 2a)。
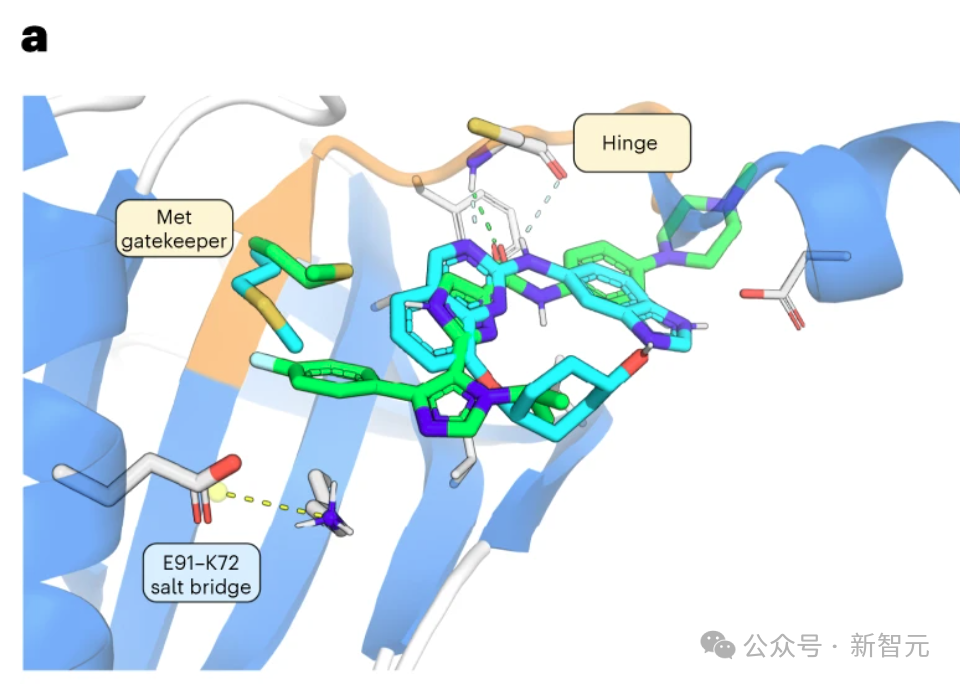
酰胺NH与近端咪唑中的氮之间的氢键作用稳定了平面构象。
此外,该咪唑的NH可以与守门员Met105的侧链形成氢键,因为NH与硫之间的距离约为 3.2 Å,适合形成氢键。
第二个咪唑将对氟苯基投射到后袋,将异丙基投射到 Asn158 和 Gln157。对氟苯基不仅被适当地容纳在后腔中,而且还与 Met105 的 C-S-C 共面(从 S 到环的最近 C 的距离为 3.8 Å)。
此前已报道了两种早期 TNIK 抑制剂 NCB-0846 和 4-甲氧基-3-(2-(3-甲氧基-4-吗啉基苯基氨基)吡啶-4-基)苯腈(化合物 9)与目标物复合物的 X 射线结构。
这些化合物与 ATP 结合位点结合,并通过两个氢键与铰链区 Cys108 的骨架相互作用(图 2a,b)。晶体结构显示,守门员 Met105 的侧链覆盖了后袋入口,因此这些化合物不包含能够转移该残基的功能。
与NCB-0846不同的是,化合物9的氰基通过靠近空腔的水分子与 Phe172 的骨架相互作用,并与 Met105 接触。
相比之下,INS018_055 被预测会深入占据后袋,在已知的 TNIK 抑制剂中形成一种独特的相互作用。为了评估 INS018_055 的结合亲和力,研究人员进行了表面等离子共振试验。与其他两种已知的 TNIK 抑制剂相比,INS018_055 显示出了强大的亲和力(Kd 值为 4.32 nM)。
INS018_055 的选择性
接下来,研究人员使用KinaseProfiler面板来评估μM INS018_055的激酶选择性。
蛋白激酶(不包括 ATM 和 DNA-PK)的活性以辐射测量的形式进行评估,而脂质激酶以及 ATM、DNA-PK 和 ATR-ATRIP(共济失调毛细血管扩张症突变和 Rad3 相关;ATR-互作蛋白)则通过均匀时间分辨荧光法进行评估。
随后,进一步评估了42个热门激酶,以确定INS018_055的IC50。
研究人员的剂量依赖性研究显示,TNIK是受抑制作用最强的靶点,其IC50为31nM。
值得注意的是,大多数在纳摩尔(<1 μM)浓度的INS018_055中达到IC50的激酶已被报道用于调节纤维化驱动过程,如ALK4、TGFBR1和DDR1。
这些结果表明,研究人员的先导化合物在纳摩尔浓度下对抑制TNIK具有选择性,并对靶向纤维化相关激酶具有更广泛的亲和力。
一项已发表的使用50个激酶面板对NCB-0846进行的选择性检测显示,有8个激酶的IC50 小于 100 nM56。
INS018_055 可抑制其中6种激酶,其IC50值大大高于该浓度。
虽然很难直接比较不同研究得出的数据(尽管两种检测方法都使用 ATP 浓度的Km值),但这些观察结果表明INS018_055具有更高的选择性(至少在已报道的激酶中是如此),这与其不同的结合模式(尤其是占据 TNIK 后袋)有关。
INS018_055 的 0 期和 I 期临床试验测试
研究人员在健康参与者中开展了一项0期单微剂量研究(ACTRN12621001541897),以评估INS018_055的药代动力学(PK),结果表明静脉注射100微克的耐受性良好。
随后,研究人员启动了一项随机、双盲、安慰剂对照的 I 期临床试验(NCT05154240),在 78 名健康志愿者中评估 INS018_055 的安全性和耐受性(首要目标)以及 PK(次要目标)。
试验的A部分是单次递增剂量给药(SAD)研究,40 名参与者按 3:1 的比例随机分配,在第 1 天按顺序剂量(10、30、60、90、120 毫克)接受 INS018_055 或安慰剂。
为了在饮食影响的背景下评估安全性和耐受性,在接受 90 毫克治疗的组群中,重复了标准高脂餐的安全性和PK评估。
B部分是一项多剂量递增给药(MAD)研究,24名健康参与者按3:1的比例随机分配,每天按顺序剂量(30、60、120毫克)服用INS015_055或安慰剂,连续服用7天。
SAD和MAD试验的PK概况
在SAD试验中,空腹个体的INS018_055血浆浓度随时间的变化曲线显示,在快速吸收阶段,INS018_055浓度达到峰值,达到最大浓度所需的中位时间(Tmax)值为服药后1.00至1.53h,随后以多相方式下降,几何平均消除半衰期(t1/2)值为7.42至9.74h。
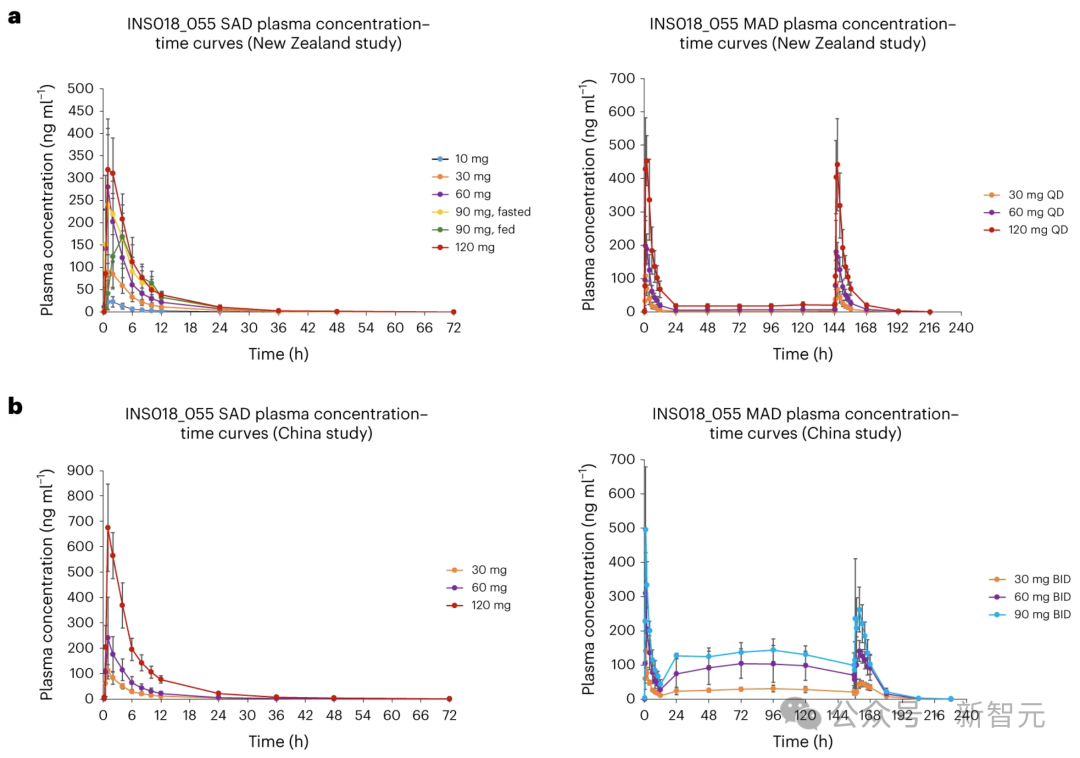
在所有剂量中,INS018_055的水平直到72小时的最后一个采样点仍可检测到,但接受30毫克剂量的组群除外,而INS018_055在服药后48小时仍可检测到。
总体而言,INS018_055的几何平均峰值(Cmax)和总暴露量(曲线下面积(AUC)0-t)以及跨时间的总暴露量(从零到无穷大的AUC(AUC0-inf))随剂量成比例增加(补充信息9),但90毫克剂量的Cmax除外,与60毫克剂量的Cmax相似。
与空腹情况(中位数Tmax为1.01小时)相比,服用90毫克的组群中,进食情况下INS018_055的血浆吸收率较低(中位数Tmax为3.00小时)。
在进食条件下,总几何Cmax和平均AUC值(Cmax 为 169 纳克/毫升-1,AUC0-t 和AUC0-inf为1390和1400小时×纳克/毫升-1)低于空腹条件下(Cmax为270纳克/毫升-1,AUC0-t和AUC0-inf为1620和1630小时×纳克/毫升-1)。
上一篇:高性能电脑配置清单
下一篇:手机屏幕自动触控问题的解决办法
产品推荐
-

售后无忧
立即购买>- DAEMON Tools Lite 10【序列号终身授权 + 中文版 + Win】
-
¥150.00
office旗舰店
-

售后无忧
立即购买>- DAEMON Tools Ultra 5【序列号终身授权 + 中文版 + Win】
-
¥198.00
office旗舰店
-

售后无忧
立即购买>- DAEMON Tools Pro 8【序列号终身授权 + 中文版 + Win】
-
¥189.00
office旗舰店
-

售后无忧
立即购买>- CorelDRAW X8 简体中文【标准版 + Win】
-
¥1788.00
office旗舰店
-
 正版软件
正版软件
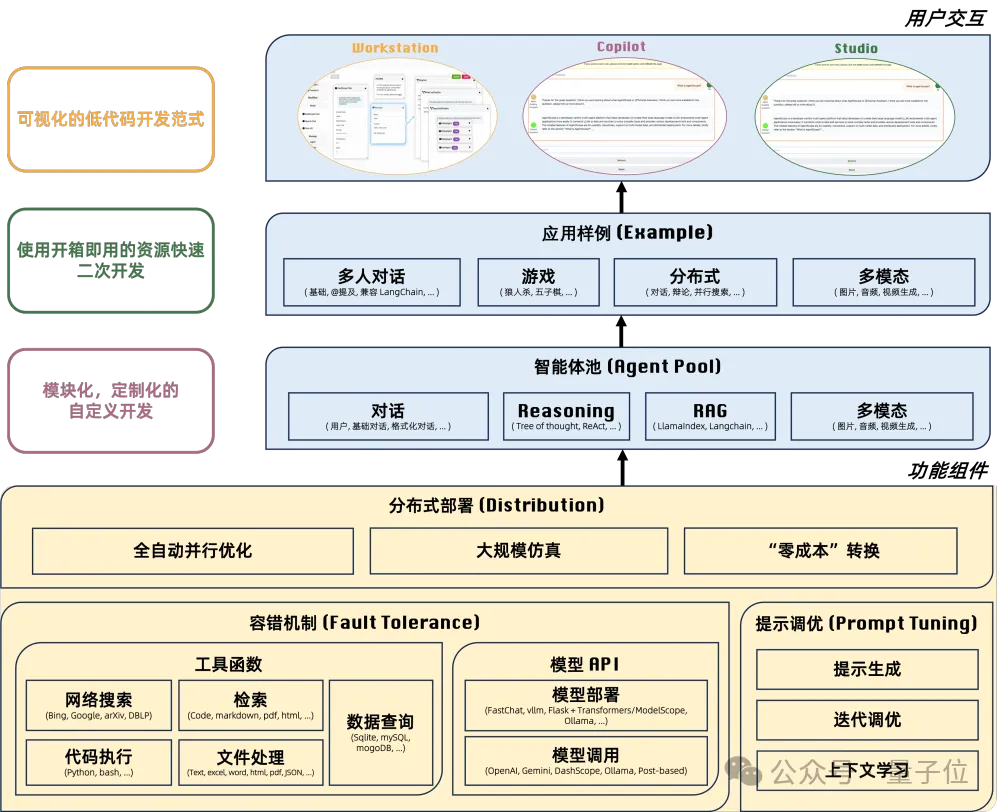
- 阿里智能体“组装工厂”开源!0经验搞定上万Agent并发
- 让多智能体开发就像搭积木,阿里巴巴通义实验室开源多智能体编程框架与开发平台AgentScope。该平台专门为多智能体应用开发者打造,旨在提供高易用的编程体验、稳定可靠的运行时保障,并且为开发者提供了分布式和多模态的技术支持。内置了OpenAI、DashScope、Gemini、Ollama等多种不同平台的模型API,深度兼容当下的大模型开源生态。AgentScope提供了多种开箱即用的功能,通过简单拖拽就能搭建多智能体应用。即使没有分布式开发经验的开发者,在AgentScope平台上也能轻松实现上万级别的
- 2分钟前 AI 开源 0
-
 正版软件
正版软件
- 胜利证券携全新APP亮相香港web3嘉年华,玩转港股、美股、虚拟币
- 2024年最大的香港web3嘉年华于4月6日开幕,胜利证券携全新APP“VictoryX”,一个集买港股、美股、虚拟货币等多项功能于一身的全能产品亮相,投资不同资产无需再频繁切换APP,一站式轻松管理各类资产,实现投资效率飞跃式的提升!胜利证券扎根香港50多年,是香港布局最广、历史最悠久的虚拟资产券商之一,见证了香港乃至全球金融市场的发展与跃迁。近年来虚拟资产的发展势不可挡,胜利证券始终走在虚拟资产交易的前沿,为投资者提供合规安全、高效透明的交易环境,以及多元化的投资机会和专业服务。一次下载,一次开户,一
- 12分钟前 0
-
 正版软件
正版软件
- 华为HDC 2024即将召开 盘古大模型5.0与鸿蒙星河版将惊艳亮相
- 华为今日正式宣布,将于6月21日至23日在东莞松山湖举办华为开发者大会(HDC2024)。备受期待的旗舰模型5.0与HarmonyOSNEXT鸿蒙星河版将在此次盛会上首次联合登场,标志着华为在AI与操作系统领域的重要突破。据悉,此次大会预计将成为华为历史上规模最宏大、吸引开发者人数最多的一次盛会。自2021年起,华为云陆续发布了包括NLP(自然语言)、CV(视觉)、科学计算在内的盘古系列预训练大模型。在去年的华为开发者大会上,华为云推出了盘古大模型3.0,深化了“AIforIndustries”战略的实施
- 27分钟前 0
-
 正版软件
正版软件
- 数字货币有哪些底层技术类型
- 数字货币采用六种主要底层技术:区块链、分布式账本技术(DLT)、哈希图、密码学、智能合约和共识机制。这些技术影响着数字货币的特性、安全性和创新可能性。
- 42分钟前 0
-
 正版软件
正版软件
- 如何基于 CKB 探索 BTC Autonomous Worlds?——Bitcoin Spring Hacker House 正式拉开序幕
- 3月24日,由NervosCKB独家赞助的BitcoinSpringHackerHouse在武汉东湖拉开序幕,持续至4月5日。此次活动吸引了超过100名独立开发者的积极报名,最终有20名优秀开发者获得了免费入住东湖风景区HackerHouse的机会,共同探索比特币和区块链技术的前沿。活动期间,3月25日至3月29日连续五天安排了一系列硬核的workshop活动,在持续五天的BTC开发课程之外,workshop内容还包括:如何深入理解闪电网络;如何基于RGB++协议表达资产所有权关系,进行花BTC资产的花式
- 57分钟前 0